DNA Damage and Mutagenesis
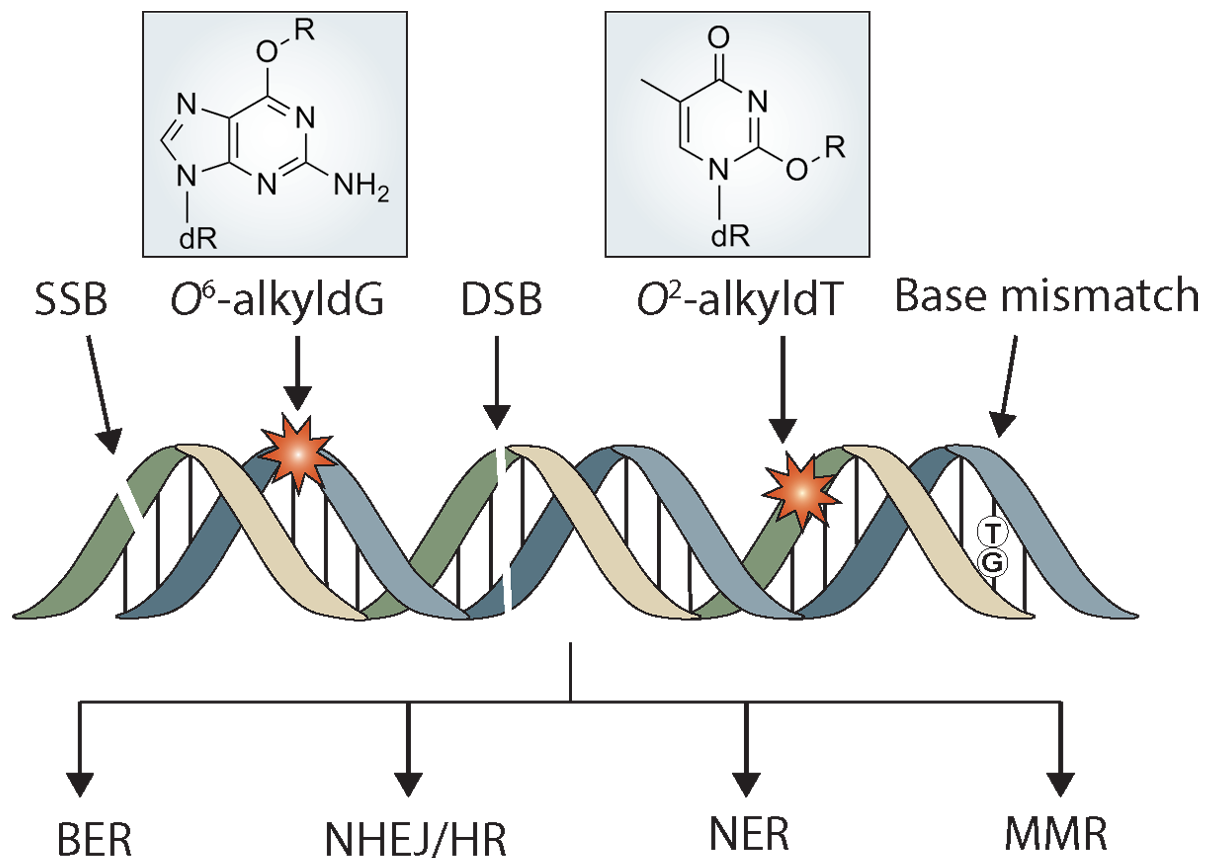
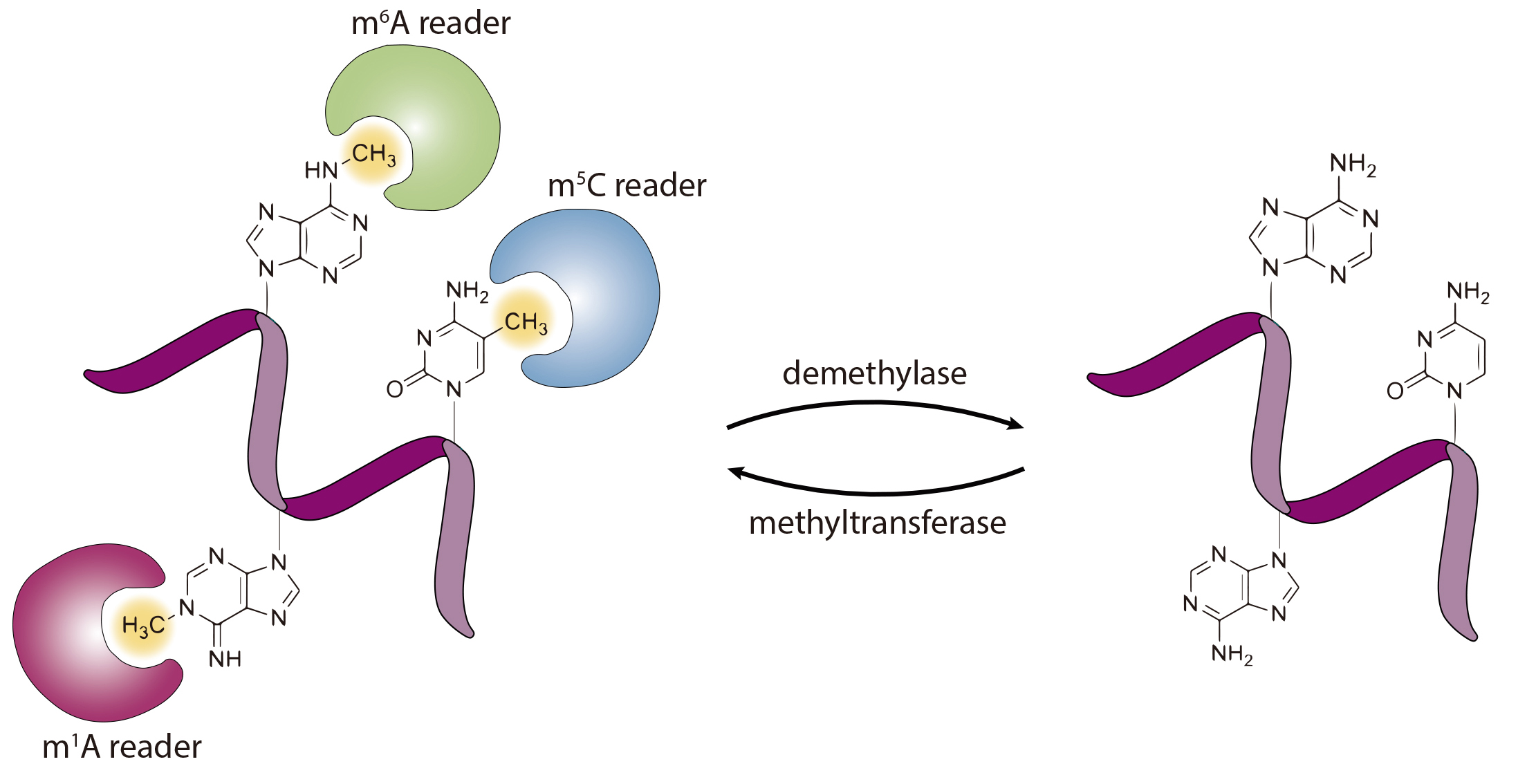
In this research area, our laboratory has been using a multi-pronged approach, encompassing mass spectrometry-based analytical chemistry, synthetic organic chemistry, biochemistry, molecular biology, and genetic tools to achieve a comprehensive understanding about the occurrene, repair and biological endpoints of DNA damage products. We aim to understand, at the molecular level, how structurally defined DNA lesions perturb the efficiency and fidelity of flow of genetic information during DNA replication and transcription. The types of DNA damage that we have been working with include DNA lesions induced by reactive oxygen species and alkylating agents. Students and postdocs working in this area are exposed to highly interdisciplinary training at the interface of chemistry and biology.